pdb2reaction Documentation¶
Version: v0.4.0 — Python CLI for enzymatic reaction-path elucidation from PDB structures using machine-learning interatomic potentials (MLIPs).
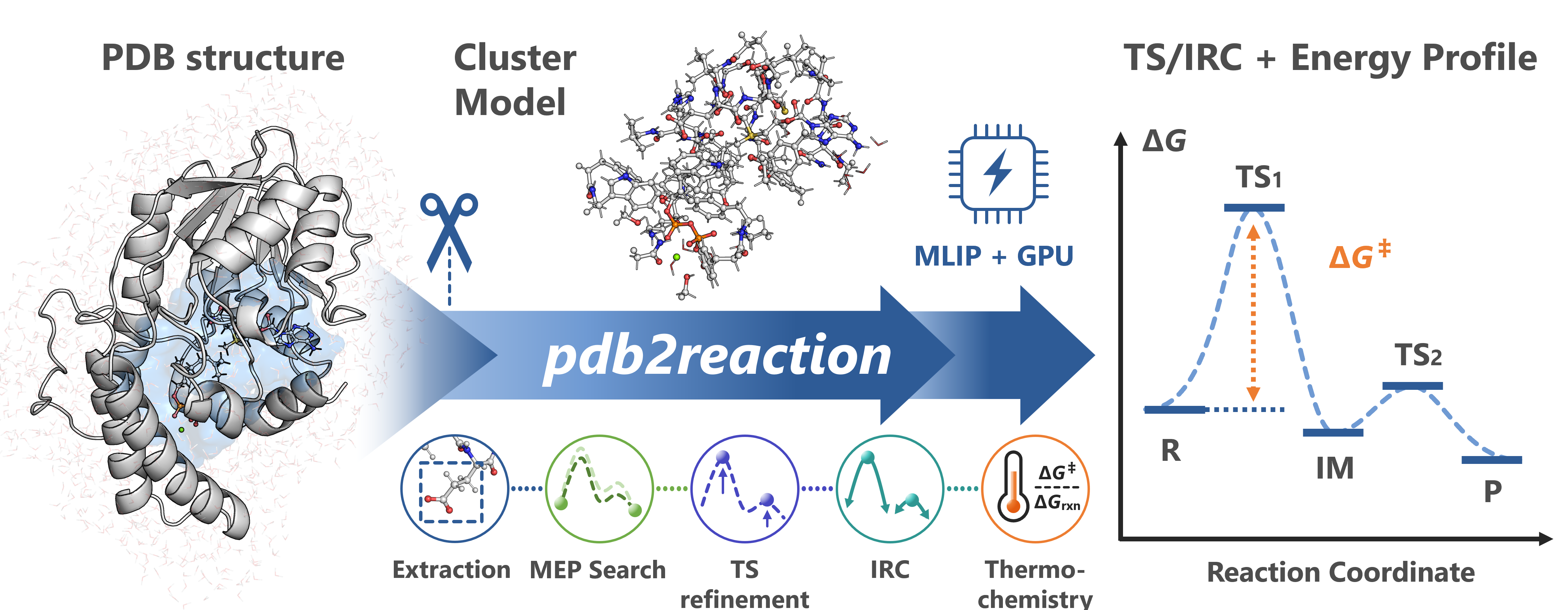
Quick start¶
Goal |
Page |
|---|---|
Install + run a first end-to-end pipeline |
|
End-to-end pipeline from a PDB |
|
Reactant only — staged distance scan |
|
TS candidate available — |
|
Choosing precision / TS route / imaginary-mode fix / controlled comparison |
|
Staged-vs-concerted scan / barrier direction |
|
Run failure / error |
|
CLI conventions / YAML / Glossary |
|
Bit-reproducible runs ( |
|
MLIP backend settings / HPC examples |
|
Cluster boundary atoms (cap H, |
Subcommands¶
Subcommand |
Description |
|---|---|
End-to-end workflow: extraction → scan → MEP → TS optimization → IRC → thermochemistry → DFT |
|
Extract active site model (binding pocket) from protein–ligand complex |
|
Resolve PDB alternate locations |
|
Repair PDB element columns (77–78) |
|
Single-structure geometry optimization (L-BFGS or RFO; optional flatten) |
|
Transition state optimization (Dimer or RS-I-RFO; optional flatten) |
|
Single-step MEP optimization via GSM or DMF (from 2 structures) |
|
Recursive multi-step MEP search with automatic refinement (2+ structures) |
|
1D bond-length driven scan with restraints |
|
2D distance grid scan |
|
3D distance grid scan |
|
Vibrational frequency analysis & thermochemistry |
|
Intrinsic Reaction Coordinate calculation |
|
Single-point DFT calculations (GPU4PySCF / PySCF) |
|
Single-point MLIP energy + forces |
|
Plot energy profiles from XYZ trajectories |
|
Draw an energy diagram from numeric values |
|
Detect and report covalent bond changes between consecutive structures |
Getting Help¶
# General help
pdb2reaction --help
# Command help
pdb2reaction <subcommand> --help
# Advanced options (dry-run, internal tuning, etc.)
pdb2reaction <subcommand> --help-advanced
Citation¶
@misc{ohmura2026pdb2reaction,
author = {Ohmura, Takuto and Sato, Hajime and Terada, Tohru},
title = {pdb2reaction: End-to-End Reaction-Path Elucidation from PDB Structures Using Machine-Learning Interatomic Potentials},
year = {2026}, doi = {10.26434/chemrxiv.15003538}, note = {ChemRxiv preprint}
}
A Zenodo software record is also available (DOI 10.5281/zenodo.19197865).
License¶
GNU General Public License v3 (GPL-3.0).