はじめに¶
概要¶
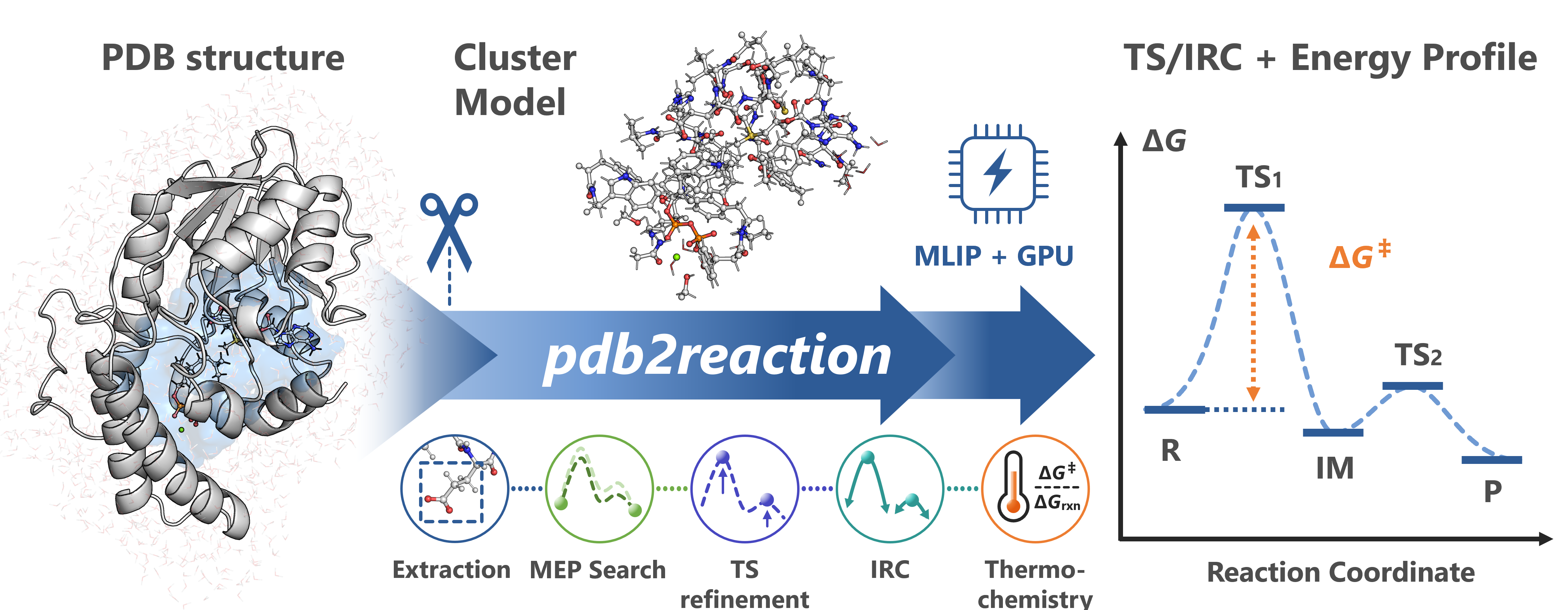
pdb2reaction は、機械学習原子間ポテンシャル(MLIP: Machine Learning Interatomic Potential)を用いて PDB 構造から酵素反応経路を解明する Python 製の CLI ツールキットです。MLIP は DFT 参照データ(エネルギー・原子間力、および周期境界条件の学習データを持つ foundation model では応力テンソルも)で学習されたニューラルネットワークモデルで、DFT のポテンシャルエネルギー曲面をごくわずかな計算コストで近似します。
多くのケースでは、次のような 1 コマンド で反応経路の初期案を得られます。
pdb2reaction -i 1.R.pdb 3.P.pdb -c 'SAM,GPP,MG' -l 'SAM:1,GPP:-3'
さらに --tsopt --thermo --dft を追加すると、最小エネルギー経路(MEP: Minimum Energy Path)探索 → 遷移状態(TS: Transition State)最適化 → 固有反応座標(IRC: Intrinsic Reaction Coordinate) → 熱化学補正 → DFT 一点計算 までまとめて実行できます。
pdb2reaction -i 1.R.pdb 3.P.pdb -c 'SAM,GPP,MG' -l 'SAM:1,GPP:-3' --tsopt --thermo --dft
実行例:
examples/ディレクトリに GPP C6-メチル基転移酵素 BezA(Tsutsumi et al., Angew. Chem. Int. Ed. 2022, 61, e202111217)の完全なallワークフロースクリプト(MEP およびスキャンパイプライン)があります。
入力として、(i) 反応順に並べたタンパク質–リガンド複合体の PDB を 2 つ以上(R → … → P)、(ii) --scan-lists/-s を指定した 1 つの PDB、または (iii) TS 候補 1 構造 + --tsopt を与えると、pdb2reaction が次を自動化します。
ユーザーが指定した基質の周辺から 活性部位モデル(バインディングポケット) を切り出し、計算用の クラスターモデル(Cluster Model)を構築
Growing String Method (GSM) や Direct Max Flux (DMF) などの経路最適化手法で 最小エネルギー経路 (MEP: Minimum Energy Path) を探索
必要に応じて 遷移状態(TS: Transition State) を最適化し、IRC(固有反応座標: Intrinsic Reaction Coordinate)計算・振動解析・DFT 一点計算 を実行
計算には機械学習原子間ポテンシャル(MLIP)を用います。デフォルトのバックエンドは Meta の UMA ですが、-b/--backend により ORB、MACE、AIMNet2 も選択できます。foundation-model 級の MLIP(UMA、ORB-v3、MACE-OMOL-0)により、クラスターモデルの TS 最適化・IRC 検証・QRRHO 熱化学が単一 GPU で実行可能となり、これまで機構スクリーニングのボトルネックだった DFT コストが大きく下がっています。想定される主な用途は以下の通りです。
DFT 等の量子化学計算では検証に時間がかかる規模の反応機構解析の試行錯誤
量子化学計算に向けた初期構造の作成(反応物・ TS ・生成物のクラスターモデル)
基質バリアントや酵素変異体にわたる反応経路の大量計算
本 CLI は最小限の手動設定で多段階の酵素反応機構を生成します。小分子系や、ユーザーが自分で構築したクラスターモデルにもそのまま適用可能です。活性部位抽出を行わない全系ワークフロー(--center/-c と --ligand-charge/-l を省略)では、.xyz / .gjf 形式の小分子(-q で正味電荷を指定)や、すでに切り出した PDB のクラスターモデルをそのまま入力できます。
HPC クラスターやマルチ GPU 環境では、UMA 推論をノード間で並列化することで、大規模なクラスターモデル(必要なら 完全なタンパク質–リガンド複合体)にも対応できます。workers と workers_per_node で並列度を設定してください(詳細は MLIP バックエンド)。
パイプライン概要¶
all サブコマンドは以下のステージを自動実行します:
PDB (R, P)
|
v
[extract] 活性部位モデル抽出(クラスターモデル)
|
v
[scan] (オプション, --scan-lists/-s)段階的距離拘束スキャン
|
v
[path-opt] MEP 探索(デフォルトは単一パス path-opt; `--refine-path True` で再帰的 path-search に切替)
|
v
[tsopt] TS 最適化 (RS-I-RFO; 代替として Dimer)
|
v
[irc] 固有反応座標
|
v
[freq] 振動解析 + 熱化学 (R, TS, P)
|
v
[dft] 一点 DFT エネルギー(オプション, --dft)
各ステージは単独のサブコマンドとしても実行できます。all はこれらを統合し、summary.json と summary.log を出力します。
主要な出力ファイル¶
ファイル |
説明 |
|---|---|
|
機械可読な結果(障壁、エネルギー、結合変化、環境情報) |
|
ディレクトリツリー付きテキストサマリ |
|
反応ステップごとの IRC 最適化 R/TS/P 構造 |
|
PyMOL/VMD で表示可能な MEP 軌跡 |
|
エネルギープロファイル図(電子/Gibbs 補正) |
Important
入力 PDB ファイルには水素原子が含まれている必要があります。
複数の PDB を提供する場合、同じ原子が同じ順序で含まれている必要があります(座標のみが異なる状態)。一致しない場合はエラーになります。
Tip
症状から切り分ける場合は、まず 典型エラー別レシピ を参照してください。 セットアップや実行でエラーに遭遇したら トラブルシューティング も参照してください。
CLI の規約¶
規約 |
例 |
備考 |
|---|---|---|
残基セレクタ |
|
複数値はシェル展開防止のためクォート |
電荷マッピング |
|
コロンで名前と電荷を区切り、カンマでエントリを区切る |
原子セレクタ |
|
区切り文字: 空白、カンマ、スラッシュ、バッククォート、バックスラッシュ |
詳細は CLI 規約 を参照してください。
水素原子付与の推奨ツール¶
PDB に水素原子がない場合は、pdb2reaction を実行する前に次のいずれかを使ってください。
ツール |
コマンド例 |
備考 |
|---|---|---|
reduce (Richardson Lab) |
|
高速、結晶構造に広く使用 |
pdb2pqr |
|
水素を追加し部分電荷を割り当て |
Open Babel |
|
汎用化学情報処理ツールキット |
PyMOL |
|
分子可視化ツール(水素付加機能あり) |
tleap (AmberTools) |
|
Amber 力場準備ツール |
複数の PDB 入力で同一の原子順序を確保するには、すべての構造に同じ水素付与ツールを一貫した設定で適用してください。
推奨クイックスタート導線¶
セットアップと依存関係の詳細は インストール を参照してください。
コマンドの基本構成¶
pip でインストールされる pdb2reaction コマンドが主な起点です。短縮エイリアス p2r も pdb2reaction パッケージが同じ setuptools entry point で登録しており(pip install pdb2reaction 直後から両方利用可能)、すべてのコマンドをどちらの名前でも実行できます。内部的には Click ライブラリを使用しており、デフォルトのサブコマンドは all です。
つまり:
pdb2reaction [OPTIONS]...
# は以下と同等
pdb2reaction all [OPTIONS]...
all は、クラスター抽出、MEP 探索、TS 最適化、振動解析、必要に応じた DFT までを 1 コマンドで一括実行するサブコマンドです。
クラスター抽出を行う場合、ワークフロー全体で共通の重要オプションが 2 つあります:
-i/--input: 1 つ以上の完全構造(反応物、中間体、生成物)-c/--center: 基質/抽出中心の定義方法(例: 残基名または残基 ID)
--center/-c を省略すると、クラスター抽出はスキップされ、完全な入力構造が直接使用されます。
主要なワークフロー¶
モード |
概要 |
クイックスタート |
|---|---|---|
複数構造 MEP(2 つ以上の PDB) |
反応座標に沿った複数の PDB(R → … → P)を受け取り、各構造のクラスターモデル抽出 → MEP 探索(デフォルトは単一パス path-opt; |
|
単一構造 + 段階的スキャン(1 PDB + |
1 つの PDB をクラスターモデル上で段階的距離スキャンにかけ、各ステージを単一パス |
|
単一構造 TSOPT のみ(1 PDB + |
MEP/経路探索を完全にスキップし、TS 候補を最適化 → 双方向 IRC → 端点最適化、必要なら R/TS/P に freq / DFT を実行 |
Important
単一入力実行には --scan-lists/-s(段階的スキャン → GSM)または --tsopt(TSOPT のみ)のいずれかが必要です。これらのいずれも指定せずに単一の -i のみを渡しても、ワークフローは実行されません。
重要な CLI オプションと動作¶
以下はワークフロー全体で最もよく使用されるオプションです。
オプション |
説明 |
|---|---|
|
入力構造。2 つ以上の PDB → MEP 探索; 1 つの PDB + |
|
基質/抽出中心を定義。残基名( |
|
電荷情報: マッピング( |
|
総電荷の強制上書き |
|
スピン多重度(例: 一重項は |
|
TS 最適化と IRC を有効化 |
|
MLIP バックエンドの選択( |
オプションの完全な一覧は CLI 規約 と 自動生成 CLI リファレンス を参照してください。
サマリーファイル¶
pdb2reaction all を実行すると、以下のサマリーファイルが出力されます。
summary.log– 結果要約summary.json– JSON 結果
主な記載内容:
実行した CLI コマンド
MEP 全体の統計(最大障壁、経路長など)
セグメントごとの障壁高さと主要な結合変化
MLIP バックエンド、熱化学、DFT 後処理で得られたエネルギー(有効な場合)
segments/seg_NN/ 配下の各セグメントディレクトリにも summary.log と summary.json があり、個別のセグメントの精密化結果を確認できます。
CLI サブコマンド¶
ほとんどのユーザは pdb2reaction all を主に使います。CLI は個別サブコマンドも提供しており、各コマンドは -h/--help に対応しています(計算/スキャン/抽出/ユーティリティ系は --help-advanced で全オプションを表示)。サブコマンド一覧と各ドキュメントへのリンクは ドキュメントトップ を参照してください。
エージェントスキル¶
pdb2reaction は skills/ に AI エージェント向けの指示書を同梱しており、CLI サブコマンド、構造 I/O(PDB / XYZ / GJF)、バックエンドインストール(UMA / Orb / MACE / AIMNet2 / DFT / xtb)、標準的なワークフロー、出力解析、HPC 運用をカバーしています。Claude Code・Codex・Cursor などのエージェントで使うには、skills/ をプロジェクト直下に(例: Claude Code なら .claude/skills/ に)または ~/.claude/skills/ にコピーしてください。
ヘルプ¶
任意のサブコマンドについて:
pdb2reaction <subcommand> --help
pdb2reaction <subcommand> --help-advanced
pdb2reaction all --help-advanced
MLIP バックエンドの詳細オプションについては MLIP バックエンド を参照してください。
問題が発生した場合は、GitHubリポジトリ で Issue を開いてください。