Getting Started¶
Overview¶
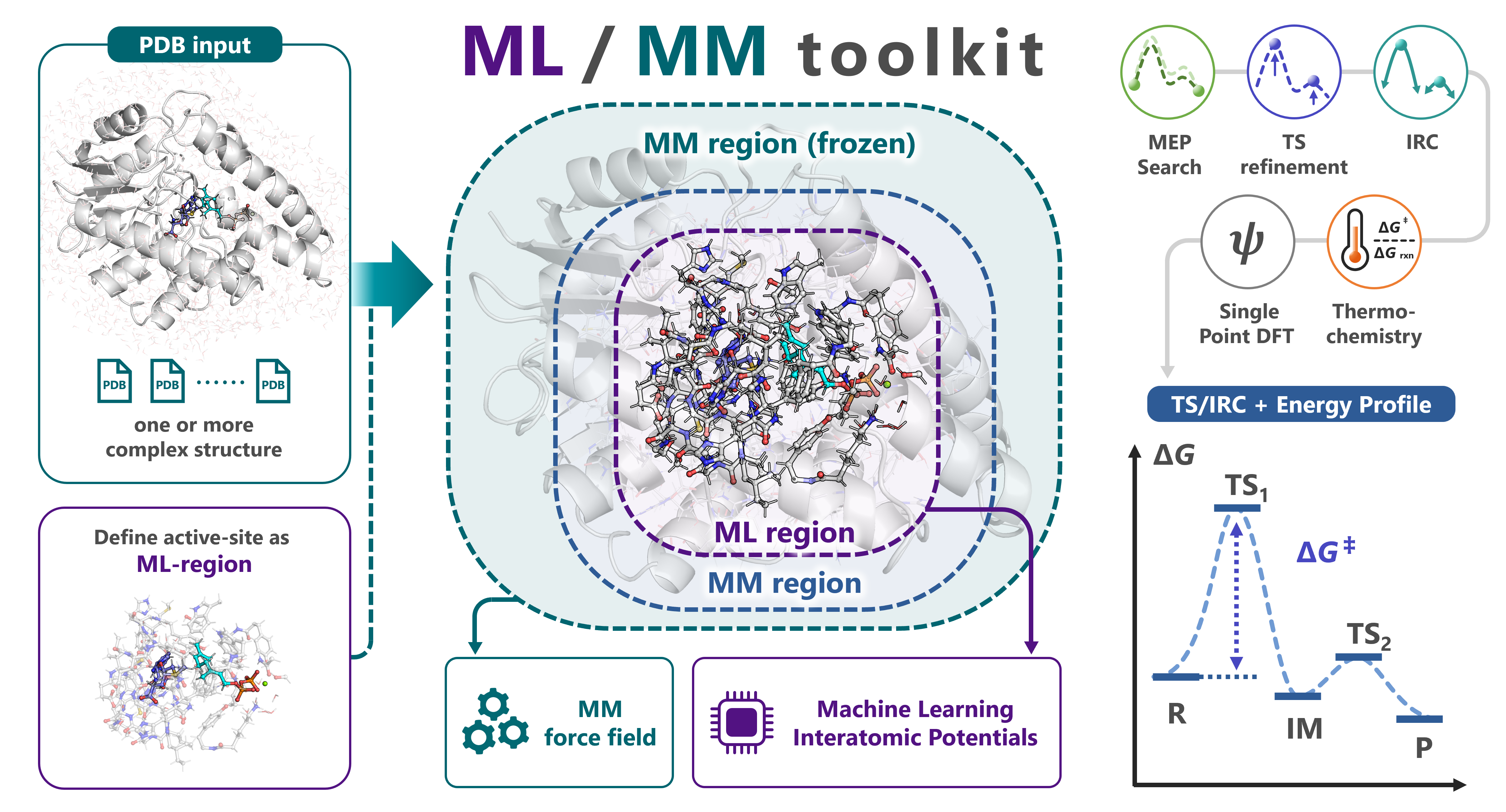
mlmm-toolkit is a Python CLI for analysing enzymatic reactions with the multi-layer ONIOM (Our own N-layered Integrated molecular Orbital and molecular Mechanics) scheme, here in an ML/MM (machine learning / molecular mechanics) variant.
Instead of the quantum-mechanical (QM) region of conventional QM/MM, it uses a machine-learning interatomic potential (MLIP) for the reactive core — default UMA, with orb / mace / aimnet2 selectable via -b. The surrounding protein is treated with mlmm-toolkit’s bundled Amber force field.
The layers are combined by the ONIOM decomposition:
E_total = E_REAL_low + E_MODEL_high - E_MODEL_low
A single command generates an initial reaction path:
mlmm all -i R.pdb P.pdb -c 'SAM,GPP' -l 'SAM:1,GPP:-3' # MEP only
mlmm all -i R.pdb P.pdb -c 'SAM,GPP' -l 'SAM:1,GPP:-3' --tsopt --thermo --dft # full
mlmm all accepts input in one of three ways:
(i) ≥ 2 PDBs (R → … → P),
(ii) one PDB with
--scan-lists, or(iii) one transition-state (TS) candidate with
--tsopt.
From that input it defines the ML region, runs mm-parm + define-layer, and performs a minimum-energy-path (MEP) search via the growing string method (GSM). It then optionally chains TS optimization, intrinsic reaction coordinate (IRC), thermochemical correction, and single-point DFT.
Important
Input PDBs must already contain hydrogen atoms. The “Input prep checklist” below covers the common pitfalls.
Multiple PDBs must share the same atoms in the same order (only coordinates differ).
Most per-stage subcommands require
--parm(frommm-parm) and--model-pdb(fromextract/define-layer);mlmm allgenerates both automatically.
For background concepts (3-layer system, link atoms, microiteration, units), read Concepts & Workflow. For symptom-first diagnosis, jump to Troubleshooting or Common Error Recipes.
CLI conventions¶
Convention |
Example |
|---|---|
Residue selector |
|
Charge mapping |
|
Atom selector |
|
Full table: CLI Conventions.
Input prep checklist¶
Hydrogens present.
mlmmdoes not auto-protonate. Add with AmberToolsreduce, OpenMMModeller.addHydrogens,pdb2pqr --ff=AMBER, Open Babelobabel -h, ormlmm mm-parm --add-h(PDBFixer wrapper). Apply the same tool to every input to keep atom order consistent.Match
-l RES:CHARGEto the H count actually in the file (e.g. SAM with 23 H =SAM:1cation, 22 H =SAM:0neutral). Mismatch breaksantechamberwith an odd-electron sqm failure — do not re-protonate “to look canonical”.R/P atom order must match. In PyMOL, tick Original atom order on export.
Multi-chain enzymes need
TERrecords between chains sotleapsegments them correctly.Per-stage subcmds:
-q/--chargeis the ML-region (ONIOM model-system) net charge, not the full-system charge. Passing the whole-enzyme charge silently builds a wrong ML region.
Installation¶
# 0. Clone the repo (skip if you only want `pip install mlmm-toolkit` once published)
git clone https://github.com/t-0hmura/mlmm_toolkit.git && cd mlmm_toolkit
# 1. New env + AmberTools + CUDA-enabled PyTorch (match your CUDA runtime)
conda create -n mlmm-toolkit python=3.11 -y && conda activate mlmm-toolkit
conda install -c conda-forge ambertools pdbfixer -y
pip install torch --index-url https://download.pytorch.org/whl/cu129
# 2. mlmm-toolkit (editable from a local clone, or `pip install mlmm-toolkit` once published)
pip install -e .
# Optional MLIP extras: pip install -e ".[orb]" / ".[aimnet]" / ".[dft]" / ".[mcp]"
# [orb] needs torch_scatter, whose prebuilt wheels live on PyG's index (not PyPI), so pip
# source-builds it and may fail with "No module named 'torch'". Add PyG's index (match torch+CUDA):
# pip install -e ".[orb]" -f https://data.pyg.org/whl/torch-2.8.0+cu129.html
# MACE: install in a dedicated env (incompatible with UMA via e3nn==0.4.4 vs >=0.5)
# 3. (UMA backend only) Authenticate Hugging Face once
# Accept the FAIR Chemistry License v1 at https://huggingface.co/facebook/UMA, then:
hf auth login # interactive
# OR: export HF_TOKEN=hf_xxx && hf auth login --token "$HF_TOKEN" --add-to-git-credential # CI / HPC
# 4. Verify
mlmm --version
Optional components¶
Component |
When to add |
Install |
|---|---|---|
|
If you see a “native extension not available” warning. JIT compilation usually handles it. |
First install |
|
Direct Max Flux (DMF) MEP backend for the standalone |
|
xTB |
|
|
Plotly Chrome |
Static PNG export beyond default |
|
HPC |
HPC clusters using environment modules |
Load before |
If you switch runtime environments (node / container / Python / PyTorch), rebuild hessian_ff in the new env. Detailed HPC job-script templates: docs/device-hpc.md.
If no usable conda xtb package is available, build xTB from source (requires GCC >= 10):
git clone --depth 1 https://github.com/grimme-lab/xtb.git
cd xtb
cmake -B build -S . -DCMAKE_BUILD_TYPE=Release
make -C build -j8
Quickstart routes¶
Quickstart:
mlmm all— multi-structure MEPQuickstart:
mlmmscan-spec route — single structure with staged bond scansQuickstart: validate TS with
mlmm tsopt— TS-only mode
Typical workflow¶
1. extract — Define ML region from full protein-ligand PDB
2. mm-parm — Generate Amber parm7/rst7 topology (requires AmberTools)
3. define-layer — Assign 3-layer ML/MM partitioning (B-factor encoding)
4. path-search — MEP search (single-pass `path-opt` by default; `--refine-path` for recursive `path-search`)
5. tsopt — Transition state optimisation
6. freq — Vibrational analysis + thermochemistry
7. dft — Single-point DFT energy refinement
mlmm all orchestrates all seven; each step is also a standalone subcommand for debugging or custom flows.
Main workflow modes¶
Mode |
Trigger |
Use when |
|---|---|---|
Multi-structure MEP |
|
You have ≥ 2 endpoints / intermediates (docking, MD, manual modeling). |
Staged scan |
|
You’d rather define reaction coordinates than provide multiple endpoints. |
TS-only |
|
You already have a TS guess and want |
mlmm [OPTIONS] is equivalent to mlmm all [OPTIONS] — all is the default subcommand, so the bare mlmm -i ... examples below run the full all workflow.
# Multi-structure MEP (richer)
mlmm -i R.pdb I1.pdb I2.pdb P.pdb -c 'SAM,GPP' -l 'SAM:1,GPP:-3' \
--out-dir ./result_all --tsopt --thermo --dft
# Staged scan
mlmm -i R.pdb -c 'SAM,GPP' -l 'SAM:1,GPP:-3' \
--scan-lists '[("TYR 285 CA","MMT 309 C10",2.20),("TYR 285 CB","MMT 309 C11",1.80)]' \
'[("TYR 285 CB","MMT 309 C11",1.20)]'
# TS-only
mlmm -i TS_CANDIDATE.pdb -c 'SAM,GPP' -l 'SAM:1,GPP:-3' --tsopt --thermo
Each tuple (i, j, target_Å) accepts a PDB atom selector ('TYR,285,CA' — space/comma/slash/backtick/backslash) or a 1-based atom index. Pass multiple stages as multiple literals after a single --scan-lists flag.
Important
Single-input runs require either --scan-lists (staged scan → GSM) or --tsopt (TS-only). A bare -i ONE.pdb will not trigger a full workflow.
Multi-backend examples¶
mlmm opt -i ml_region.pdb --parm real.parm7 --model-pdb ml.pdb -q 0 -b orb # ORB
mlmm opt -i ml_region.pdb --parm real.parm7 --model-pdb ml.pdb -q 0 -b mace # MACE
mlmm opt -i ml_region.pdb --parm real.parm7 --model-pdb ml.pdb -q 0 --embedcharge # xTB embedding
DFT refinement via Gaussian / ORCA (hand-off)¶
mlmm-toolkit provides a round-trip hand-off — Gaussian or ORCA must be licensed and on PATH separately:
# 1. ML/MM TS refinement
mlmm tsopt -i ts_guess.pdb --parm real.parm7 --model-pdb ml_region.pdb -q 0 -m 1
# 2. Export to Gaussian ONIOM (.com)
mlmm oniom-export --mode g16 --parm real.parm7 -i result_tsopt/final_geometry.pdb \
--model-pdb ml_region.pdb -o ts_refine.com -q 0 -m 1 --method "wB97XD/def2-TZVPD"
# 3. Run externally (ORCA via --mode orca also supported)
g16 < ts_refine.com > ts_refine.log
# 4. Pull DFT-refined geometry back in
mlmm oniom-import -i ts_refine.com --ref-pdb ml_region.pdb -o ts_dft
# 5. Continue freq / IRC on the refined geometry
mlmm freq -i ts_dft_layered.pdb --parm real.parm7 --model-pdb ml_region.pdb -q 0 -m 1
mlmm irc -i ts_dft_layered.pdb --parm real.parm7 --model-pdb ml_region.pdb -q 0 -m 1
Full flag references: oniom-export, oniom-import, oniom-gaussian, oniom-orca.
Common options¶
Option |
Description |
|---|---|
|
Input structures. See the “Main workflow modes” table above for how the input count and accompanying flags select a mode. |
|
Substrate / extraction center (residue names |
|
Charge mapping ( |
|
ML-region net charge / spin multiplicity. |
|
Staged distance scans for single-input runs (literals or YAML/JSON file). |
|
Top-level output directory. |
|
TS optimization + IRC / vibrational analysis / single-point DFT. |
|
Single-pass |
|
MLIP backend (default |
|
xTB point-charge embedding correction (default off). |
|
ML Hessian mode. |
Direct Max Flux (DMF) MEP is selectable only via the standalone path-search / path-opt subcommands (--mep-mode dmf). mlmm all always uses GSM, so passing --mep-mode to mlmm all is silently ignored.
Full option matrix and YAML schema: YAML Reference. Subcommand-by-subcommand table: README “CLI Subcommands”.
Run summaries¶
Every mlmm all run writes summary.log (human) + summary.json (machine) with the CLI command, global MEP statistics, per-segment barriers / bond changes, and MLIP / thermo / DFT energies (when enabled). Per-segment segments/seg_NN/ subdirectories carry their own summaries.
Getting help¶
mlmm --help # top-level
mlmm <subcommand> --help # core options
mlmm <subcommand> --help-advanced # full option set
Driving from an AI coding agent¶
mlmm-toolkit ships skills/ with agent-readable instructions. Copy skills/ into your project as .claude/skills/ (or merge into ~/.claude/skills/) for Claude Code / Cursor / OpenCode pickup.
Warning
This software is still under development. Please use it at your own risk.