mlmm-toolkit Documentation¶
Version: v0.3.0 — Python CLI for ML/MM ONIOM analyses of enzymatic reactions.
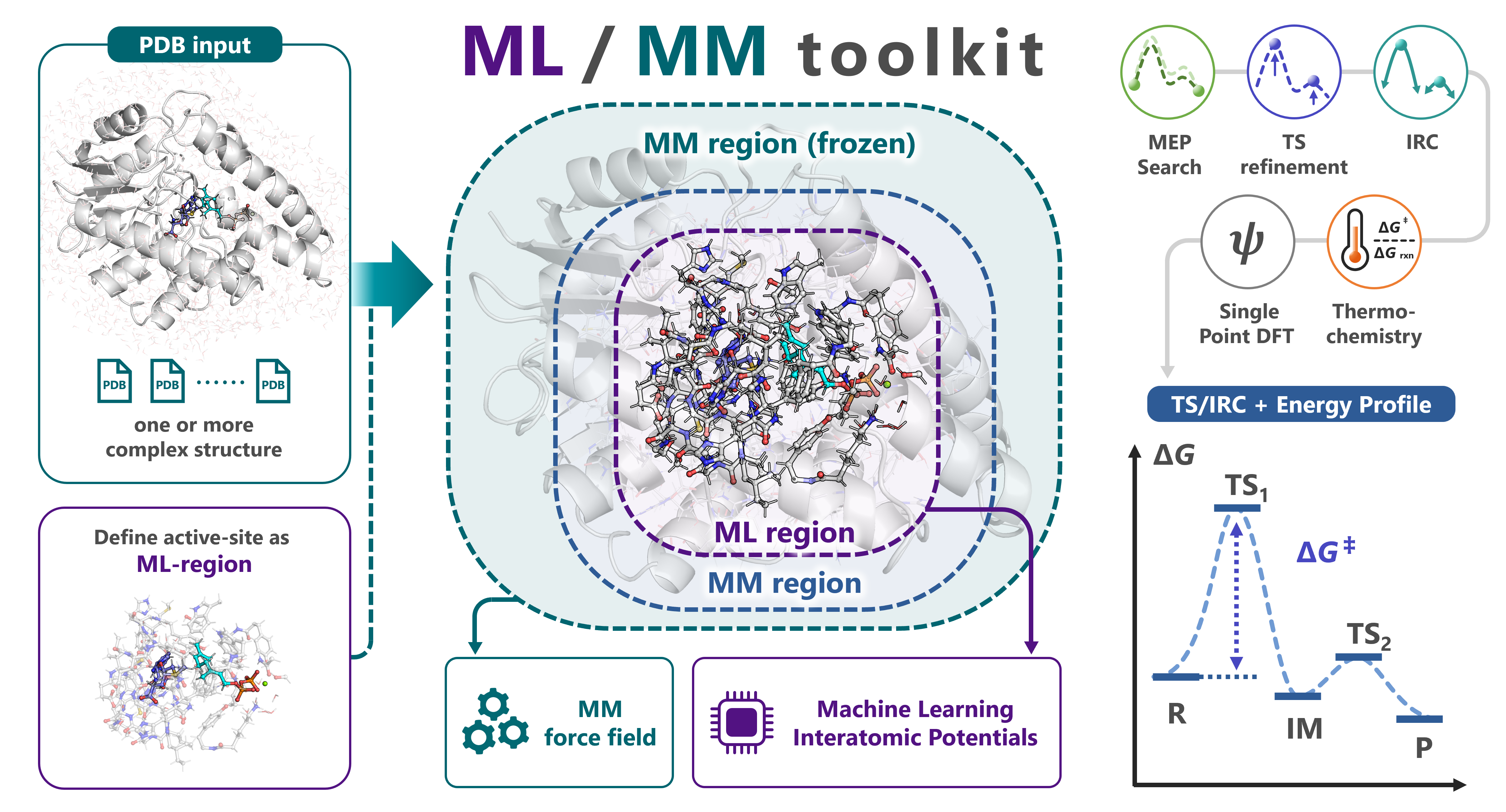
Quick start¶
Goal |
Page |
|---|---|
Install + run a first end-to-end pipeline |
|
3-layer ONIOM, microiteration, link atoms |
|
End-to-end pipeline from a PDB |
|
Single-structure staged scan |
|
TS validation ( |
|
TS routes, imaginary-frequency fixes, controlled mutant comparison |
|
Scan modes (staged vs concerted), barrier direction |
|
Precision by GPU class |
|
Symptom-first failure routing |
|
CLI conventions, YAML schema |
|
Python API / ML/MM calculator architecture |
|
Configure GPU/CPU devices and submit on HPC |
|
Terminology |
Subcommands¶
Subcommand |
Role |
|---|---|
End-to-end: ML/MM model setup → MEP → TS → IRC → freq → DFT |
|
|
Structure preparation |
Geometry / TS optimization |
|
MEP optimization / recursive refinement |
|
1D / 2D / 3D bond-distance scans |
|
Vibrational analysis + thermochemistry / IRC (EulerPC) |
|
Single-point DFT / single-point ML/MM ONIOM |
|
Bond-change report between consecutive structures |
|
Energy plot / R→TS→P diagram |
|
Gaussian / ORCA QM/MM round-trip |
Citation¶
Ohmura, T., Inoue, S., Terada, T. (2025). ML/MM toolkit — Towards Accelerated Mechanistic Investigation of Enzymatic Reactions. ChemRxiv. https://doi.org/10.26434/chemrxiv-2025-jft1k
License¶
GNU General Public License v3 (GPL-3.0).